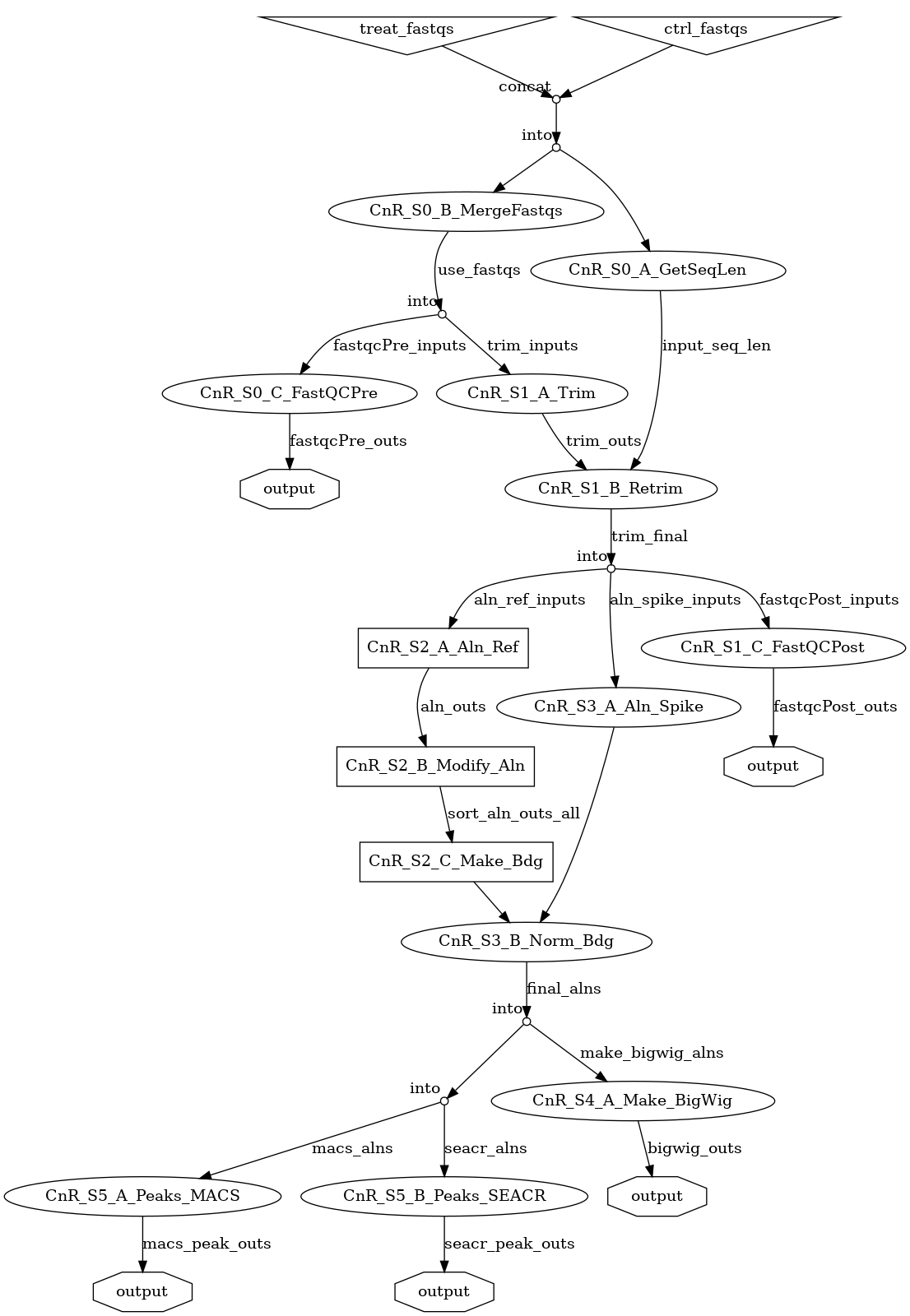
Workflow¶
- Full Workflow:
Download and Installation¶
(If necessary, begin by installing Nextflow and Conda as described in Quickstart)
Nextflow allows the pipeline to be downloaded simply by using the “nextflow run” command, and providing the user/repository path to the relevant github repository. CnR-flow has a one-step installation mode that can be performed simultaneously (--mode initiate).Together, this gives the command:$ nextflow run RenneLab/CnR-flow --mode initiateThis should download the pipeline, install the few required local files and dependencies, and prepare the pipeline for execution.Note
After the initial download, the pipeline can then be referred to by name, as with:“nextflow run CnR-flow …”
Dependency Setup and Validation¶
CUT&RUN-Flow comes preconfigured to utilize the Conda package management suite together with Bioconda packages to handle pipeline dependencies (currently supported with Linux64 systems, with macOS support under development).For even greater reproducibility, it is now recommended to utilize Docker (`-profile docker`) or Singularity (`-profile singularity`) to execute pipeline steps where these options are available.for more details, or for an alternative configuration, see Dependency ConfigOnce dependency setup has been completed, it can be tested using the--mode validaterun mode.# validate only steps enabled in nextflow.config $ nextflow run CnR-flow --mode validate
Reference Preparation¶
CnR-flow provides one-step preparation of alignment reference genome(s) for use by the pipeline. Either the local path or URL of a fasta file are provided to the--ref_fasta (params.ref_fasta)paramater and the execution is performed with:$ nextflow run CnR-flow --mode prep_fastaThis copies the reference fasta to the directory specified by--ref_dir (params.ref_dir), creates a bowtie2 alignment reference, creates a fasta index using Samtools, and creates a “.chrom.sizes” file using UCSC faCount. The effective genome size is also calculated with faCount, using the (Total - N’s) method. [faCount_Citation] Reference details are written to a “.refinfo.txt” in the same directory.Note
If spike-in normalization is enabled, the same process will be repeated for the fasta file supplied to
--norm_ref_fasta (params.norm_ref_fasta)for alignments to the spike-in control genome.These referenes are then detected automatically, using the same parameter used for preparation setup. For more details, see Reference Files Setup. The list of all detectable prepared databases can be provided using the--mode list_refsrun mode:$ nextflow run CnR-flow --mode list_refs
Experimental Condition¶
CUT&RUN-Flow allows automated handling of treatment (Ex: H3K4me3) and and control (Ex: IgG) input files, performing the analysis steps on each condition in parallel, and then associating the treatment with the control for the final peak calling step. This can be performed either with a single treatment/control group, or with multiple groups in parallel. For more information, see Task Setup.
Preprocessing Steps¶
GetSeqLen¶
This step is enabled with paramater
--do_retrim (params.do_retrim)(default:true). This step takes one example input fastq[.gz] file and determines the sequence length, for use in later steps.
MergeFastqs¶
This step is enabled with paramater
--do_merge_lanes (params.do_merge_lanes)(default:true). If multiple sets of paired end files are provided that differ only by the “_L00#_” component of the name, these sequences are concatenated for further analysis.For example, these files will be merged into the common name: ‘my_sample_CTRL_R(1/2)_001.fastq.gz’:
./my_sample_CTRL_L001_R1_001.fastq.gz ./my_sample_CTRL_L001_R2_001.fastq.gz ./my_sample_CTRL_L002_R1_001.fastq.gz ./my_sample_CTRL_L002_R2_001.fastq.gz #... --> ./my_sample_CTRL_R1_001.fastq.gz ./my_sample_CTRL_R2_001.fastq.gz
FastQCPre¶
This step is enabled with paramater
--do_fastqc (params.do_fastqc)(default:true). FastQC is utilized to perform quality control checks on the input (presumably untrimmed) fastq[.gz] files. [FastQC_Citation]
Trim¶
This step is enabled with paramater--do_trim (params.do_trim)(default:true). Trimming of input fastq[.gz] files for read quality and adapter content is performed by Trimmomatic. [Trimmomatic_Citation]Default trimming parameters:trimmomatic_adapter_mode = "ILLUMINACLIP:" trimmomatic_adapter_params = ":2:15:4:4:true" trimmomatic_settings = "LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 MINLEN:25"Default flags:trimmomatic_flags = "-phred33"
FastQCPost¶
This step is enabled with paramater
--do_fastqc (params.do_fastqc)(default:true). FastQC is utilized to perform quality control checks on sequences after any/all trimming steps are performed. [FastQC_Citation]
Alignment Steps¶
Aln_Ref¶
Sequence reads are aligned to the reference genome using Bowtie2. [Bowtie2_Citation]Default alignment parameters were selected using concepts presented in work by the Henikoff Lab [Meers2019] and the Yuan Lab [CUTRUNTools_Citation].Default flags:aln_ref_flags = "--local --very-sensitive-local --phred33 -I 10 -X 700 --dovetail --no-unal --no-mixed --no-discordant"Warning
None of the output alignments (.sam/.bam/.cram) files produced in this step (or indeed, anywhere else in the pipeline) are normalized. The only normalized outputs are genome coverage tracks produced if normalization is enabled.
Modify_Aln¶
Output alignments are then subjected to several cleaning, filtering, and preprocessing steps utilizing Samtools. [Samtools_Citation]These are:
Removal of unmapped reads (samtools view)
Sorting by name (samtools sort [required for fixmate])
Adding/correcting mate pair information (samtools fixmate -m)
Sorting by genome coordinate (samtools sort)
Marking duplicates (samtools mkdup)
( Optional Processing Steps [ see below ] )
Alignment compression BAM -> CRAM (samtools view)
Alignment indexing (samtools index)
Optional processing steps include:
Removal of Duplicates
Filtering to reads <= 120 bp in length
The desired category (or categories) of output are selected with--use_aln_modes (params.use_aln_modes). Multiple categores can be specifically selected usingparams.use_aln_modesas a list, and the resulting selections are analyzed and output in parallel.(Example:params.use_aln_modes = ['all', 'less_120_dedup'])
Option
Deduplicated
Length <= 120bp
all
false
false
all_dedup
true
false
less_120
false
true
less_120_dedup
true
true
Default mode:use_aln_modes = ["all"] // Options: ["all", "all_dedup", "less_120", "less_120_dedup"]
Make_Bdg¶
Further cleaning steps are then performed on the outputs, to prepare the alignments for (optional) normalization and peak calling.These modifications are performed as suggested by the Henikoff lab in the documentation for SEACR, https://github.com/FredHutch/SEACR/blob/master/README.md [SEACR_Citation] , and are performed utilizing Samtools [Samtools_Citation] and bedtools [bedtools_Citation].These are:
Sorting by name and uncompress CRAM -> BAM (samtools sort)
Covert BAM to bedgraph (bedtools bamtobed)
Filter discordant tag pairs (awk)
Change bedtools bed format to BEDPE format (cut | sort)
Convert BEDPE to (non-normalized) bedgraph (bedtools genomecov)
Note
Genome coverage tracks output by this step are NOT normalized.
Normalization Steps¶
Aln_Spike¶
This step is enabled with paramater--do_norm_spike (params.do_norm_spike)(default:true).This step calculates a normalization factor for scaling output genome coverage tracks.
- Strategy:
A dual-alignment strategy is used to filter out any reads that cross-map to both the primary reference and the normalization references. Sequence pairs that align to the normalization reference are then re-aligned to the primary reference. The number of read pairs that align to both references is then subtracted from the normalization factor output by this step, depending on the value of
--norm_mode (params.norm_mode)(default:true).- Details:
Default flags:
aln_norm_flags = params.aln_ref_flagsAll reads that aligned to the normalization reference are then again aligned to the primary reference using Bowtie2. [Bowtie2_Citation]Counts are then performed of pairs of sequence reads that align (and re-align, respectively) to each reference using Samtools (viasamtools view). [Samtools_Citation] The count of aligned pairs to the spike-in genome reference is then returned, with the number of cross-mapped pairs subtracted depending on the value of--norm_mode (params.norm_mode).
norm_mode
Normalization Factor Used
all
norm_ref_aligned (pairs)
adj
norm_ref_aligned - cross_map_aligned (pairs)
norm_mode = 'adj' // Options: ['adj', 'all'] seacr_norm_mode = "auto" // Options: "auto", "norm", "non"
Norm_Bdg¶
This step is enabled with paramater--do_norm_spike (params.do_norm_spike)(default:true).This step uses a normalization factor to create scaled genome coverage tracks. The calculation as provided by the Henikoff Lab: https://github.com/Henikoff/Cut-and-Run/blob/master/spike_in_calibration.csh [Meers2019] is:\(count_{norm} = (count_{site} * norm\_scale) / norm\_factor\)
Thus, the scaling factor used is calucated as:\(scale\_factor = norm\_scale / norm\_factor\)
Wherenorm_factoris calculated in the previous step, and the arbitrarynorm_scaleis provided by the parameter:--norm_scale (params.norm_scale).Defaultnorm_scalevalue:norm_scale = 1000 // Arbitrary value for scaling of normalized counts.The normalized genome coverage track is then created by bedtools using the-scaleoption. [bedtools_Citation]
Aln_CPM¶
(Beta Implementation)This step is enabled with paramater--do_norm_cpm (params.do_norm_cpm)(default:false).As an alternative option to spike-in normalization, this “counts-per-million” normalization method calculates a normalization factor for scaling output genome coverage tracks by dividng by the total number of reference-aligned tags per sample, multiplying by 1M, and then by an arbitrary scaling factor.Counts of reference-aligned reads are made using using Samtools (viasamtools view). [Samtools_Citation]The per-site scaled count is then calculated:\(count_{norm} = (count_{site} * norm\_scale) 1,000,000 / total\_ref\_aln\_pairs\)
Thus, the scaling factor used is calucated as:\(scale\_factor = norm\_scale * 1,000,000 / total\_ref\_aln\_pairs\)
The arbitrarynorm_cpm_scaleis provided by the parameter:--norm_cpm_scale (params.norm_cpm_scale).Defaultnorm_cpm_scalevalue:norm_cpm_scale = 1000 // Arbitrary value for scaling of normalized counts.The normalized genome coverage track is then created by bedtools using the-scaleoption. [bedtools_Citation]
Conversion Steps¶
Make_BigWig¶
This step is enabled with paramater--do_make_bigwig (params.do_make_bigwig)(default:true).This step converts the output genome coverage file from the previous steps as in the UCSC bigWig file format using UCSC bedGraphToBigWig, a genome coverage format with significantly decreased file size. [bedGraphToBigWig_Citation]Warning
The bigWig file format is a “lossy” file format that cannot be reconverted to bedGraph with all information intact.
Peak Calling Steps¶
--peak_callers (params.peak_callers).
This can either be provided a single argument, as with:
--peak_callers seacr (params.peak_callers = "seacr")
params.peak-callers = ['macs', 'seacr']
peak_callers value:peak_callers = ['macs', 'seacr'] // Options: ['macs', 'seacr']
Peaks_MACS2¶
This step is enabled ifmacsis included inparams.peak-callers.This step calls peaks using the non-normalized alignment data produced in previous steps, using the MACS2 peak_caller. [MACS2_Citation]Default MACS2 Settings:
// Macs2 Settings macs_qval = '0.01' macs_flags = ''
Peaks_SEACR¶
This step is enabled ifseacris included inparams.peak_callers.This step calls peaks using the normalized alignment data produced in previous steps (if normalization is enabled, using the SEACR peak caller. [SEACR_Citation]Special thanks to Michael Meers and the Henikoff Group for their permission to distribute SEACR bundled with this pipeline.
- Parameters:
--seacr_norm_mode (params.seacr_norm_mode)passes eithernormornonto SEACR. Options:
'auto':
if
do_norm = true- Passes'non'to SEACRif
do_norm = false- passes'norm'to SEACR
'norm'
'non'--seacr_fdr (params.seacr_fdr)is passed directly to SEACR.--seacr_call_stringent (params.seacr_call_stringent)- SEACR is called in “stringent” mode.--seacr_call_relaxed (params.seacr_call_relaxed)- SEACR is called in “relaxed” mode.(If both of these are true, both outputs will be produced)Default SEACR Settings:
// SEACR Settings seacr_fdr_threshhold = "0.01" seacr_norm_mode = "auto" // Options: "auto", "norm", "non" seacr_call_stringent = true seacr_call_relaxed = true