CUT&RUN-Flow (CnR-flow)¶



- Pipeline Design:
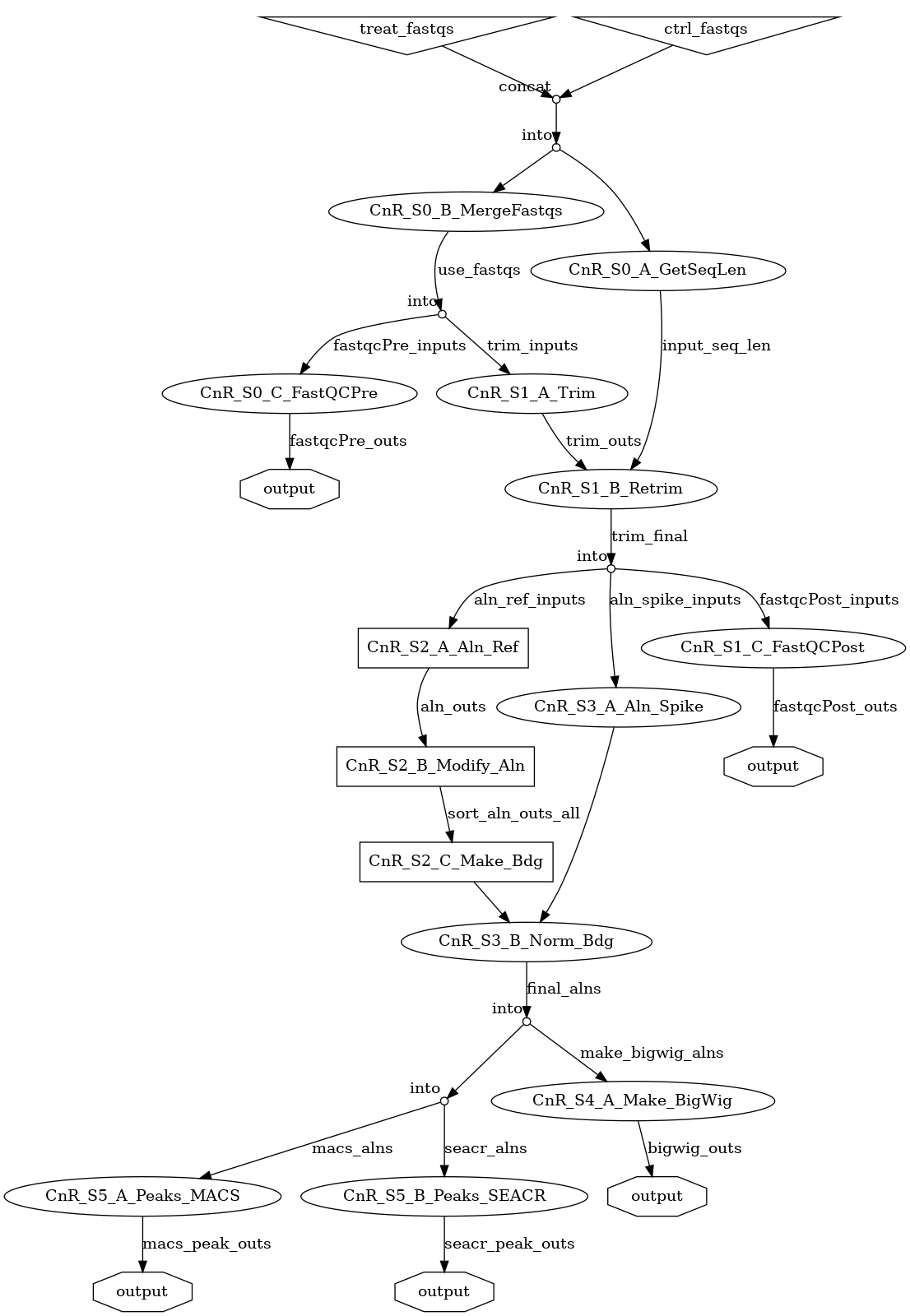
- CUT&RUN-Flow is built using Nextflow, a powerful domain-specific workflow language built to create flexible and efficient bioinformatics pipelines. Nextflow provides extensive flexibility in utilizing cluster computing environments such as PBS and SLURM, and in automated and compartmentalized handling of dependencies using Conda / Bioconda, Docker, Singularity or Environment Modules.
- Dependencies:
- In addition to local configurations, Nextflow handles dependencies in separated working environments within the same pipeline using Conda or Environment Modules within your working environment, or using container-encapsulated execution with Docker or Singularity. CnR-flow is pre-configured to auto-acquire dependencies with no additional setup, either using Conda recipes from the Bioconda project, or by using Docker or Singularity to execute Docker images hosted by the BioContainers project (Bioconda; BioContainers).CUT&RUN-Flow utilizes UCSC Genome Browser Tools and Samtools for reference library preparation, FastQC for tag quality control, Trimmomatic for tag trimming, Bowtie2 for tag alignment, Samtools, bedtools and UCSC Genome Browser Tools for alignment manipulation, and MACS2 and/or SEACR for peak calling, as well as their associated language subdependencies of Java, Python2/3, R, and C++.
- Pipeline Features:
One-step reference database prepration using a path (or URL) to a FASTA file.
Ability to specify groups of samples containing both treatment (Ex: H3K4me3) and control (Ex: IgG) antibody groups, with automated association of each control sample with the respective treatment samples during the peak calling step
Built-in normalization protocol to normalize to a sequence library of the user’s choice when spike-in DNA is used in the CUT&RUN Protocol (Optional, includes an E. coli reference genome for utiliziation of E. coli as a spike-in control as described by Meers et. al. (eLife 2019) [see the References section of this documentation])
OR: CPM-normalization to normalize total read counts between samples (beta).
SLURM, PBS… and many other job scheduling environments enabled natively by Nextflow
Output of memory-efficient CRAM (alignment), bedgraph (genome coverage), and bigWig (genome coverage) file formats
- Quickstart:
Here is a brief introduction on how to install and get started using the pipeline. For full details, see this documentation.
- Prepare Task Directory:
- Create a task directory, and navigate to it.
$ mkdir ./my_task # (Example) $ cd ./my_task # (Example)
- Install Nextflow (if necessary):
- Download the nextflow executable to your current directory.(You can move the nextflow executable and add to $PATH for future usage)
$ curl -s https://get.nextflow.io | bash # For the following steps, use: nextflow # If nextflow executable on $PATH (assumed) ./nextflow # If running nextflow executable from local directory
- Download and Install CnR-flow:
- Nextflow will download and store the pipeline in the user’s Nextflow info directory (Default:
~/.nextflow/)$ nextflow run RenneLab/CnR-flow --mode initiate
- Configure, Validate, and Test:
- Conda:
Install miniconda (if necessary). Installation instructions
The CnR-flow configuration with Conda should then work “out-of-the-box.”
- Docker:
Add ‘-profile docker’ to all nextflow commands
- Singularity:
Add ‘-profile singularity’ to all nextflow commands
If using an alternative configuration, see the Dependency Config section of this documentation for dependency configuration options.Once dependencies have been configured, validate all dependencies:# Conda or other configs: $ nextflow run CnR-flow --mode validate_all # OR Docker Configuration: $ nextflow run CnR-flow -profile docker --mode validate_all # OR Singularity Configuration: $ nextflow run CnR-flow -profile singularity --mode validate_all
Fill the required task input parameters in “nextflow.config” For detailed setup instructions, see the Task Setup section of this documentation Additionally, for usage on a SLURM, PBS, or other cluster systems, configure your system executor, time, and memory settings.# Configure: $ <vim/nano...> nextflow.config # Task Input, Steps, etc. Configuration #REQUIRED values to enter (all others *should* work as default): # ref_fasta (or some other ref-mode/location) # treat_fastqs (input paired-end fastq[.gz] file paths) # [OR fastq_groups] (mutli-group input paired-end .fastq[.gz] file paths)
- Prepare and Execute Pipeline:
- Prepare your reference databse (and normalization reference) from .fasta[.gz] file(s):
$ nextflow run CnR-flow --mode prep_fasta
Perform a test run to check inputs, paramater setup, and process execution:$ nextflow run CnR-flow --mode dry_run
If satisifed with the pipeline setup, execute the pipeline:$ nextflow run CnR-flow --mode run
Further documentation on CUT&RUN-Flow components, setup, and usage can be found in this documentation.
Documentation Contents: